IR-Binder-000001 annotations now include the full input sequence to facilitate data re-use. Sequences were exported with -targetSequences in MiXCR v 3.0.8. The original dataset from 2020-07-02 did not include input sequences. The original data associated with IR-Binder-000001 has been extended to include the full sequence which was left out of the original data load (using MiXCR with -targetSequences).
Consistent with the “living repository ” approach described by Schultheiß et al. –, a repository that is continuously fed with new annotated sequence data – a new data cohort has been added (Study ID IR-Binder-000002). It comprises 24 TRB repertoires from 14 subjects.
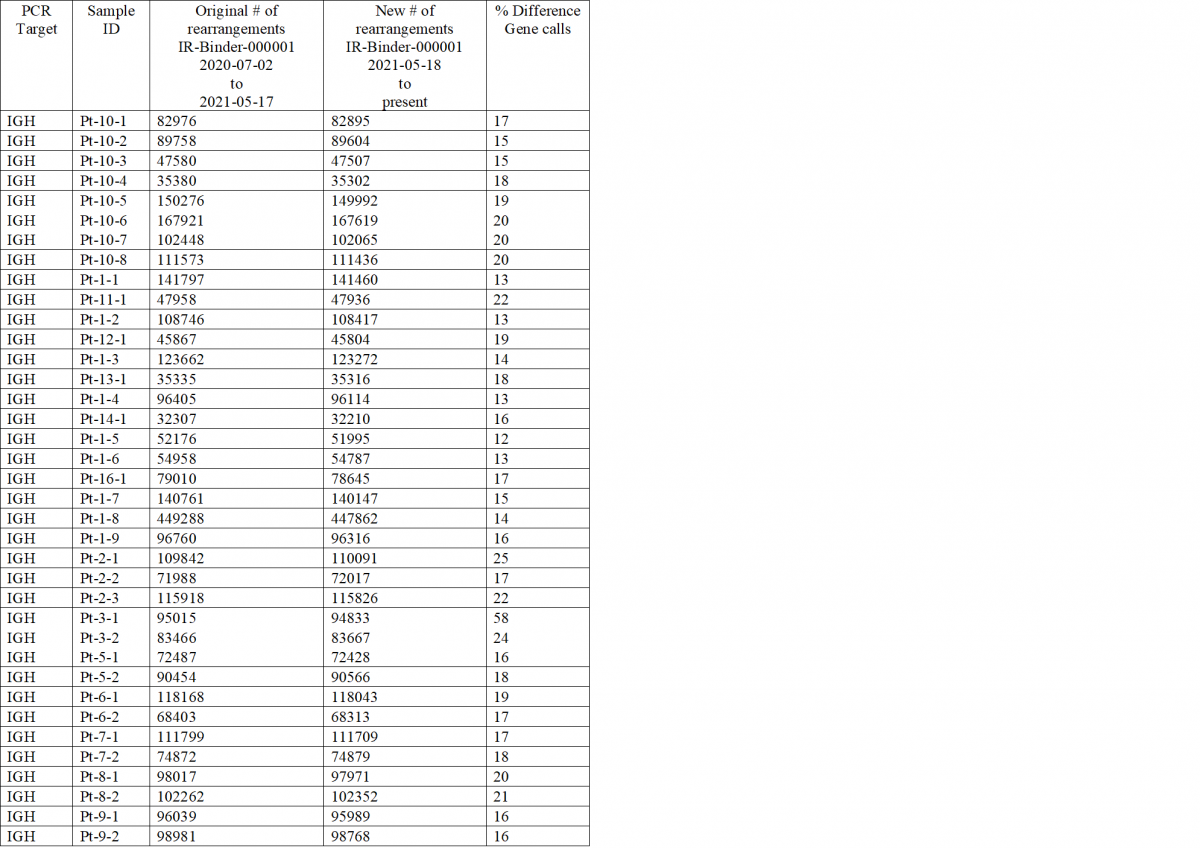
Special Notes: When the authors re-annotated IR-Binder-000001 to obtain full sequences, the annotations of some IGH samples changed in terms of both number of sequences and gene calls; these differences were approved by authors. Data accessed through the iReceptor Gateway prior to 2021-05-18 would have retrieved the original annotations, data accessed after 2021-05-18 will retrieve the new annotations. The table below documents affected samples (total # of sequences and % of annotations with different gene calls (V, D, and/or J), relative to the original file). The % difference ranges from 12-58% (average = 18%). For sample Pt-3-1 (58% difference), the majority of difference (87%) was due to IGHV1-69*13 calls being changed to IGHV1-69*01. All other samples appeared less dominated by IGHV1-69 in terms of both gene calls and gene calls changed. These differences likely arose from the challenge of computationally assigning highly variable sequence regions (having undergone somatic hypermutation) to highly similar IGHV1-69 reference alleles (only 2 SNPs differentiate *01, *12, and *13).
The original data associated with IR-Efimov-000001 has been extended to include the full sequence which was left out of the original data load (using MiXCR with -targetSequences).
Some samples/repertoires were removed for subjects p1449 (3) and p1489 (2). These include sample IDs (sample_id):
Some samples/repertoires differ in the number of rearrangements:
p1437_PBMC (old = 3949, new = 127996)
p1484_CD8ifny (old = 66, new = 237)
p1484_CD4ifny (old = 5755, new = 89)
p1489_YLQ_neg_alpha (old = 89351, new = 27966)
p1489_YLQ_neg_beta p(old = 133371, new = 20751)
Updates to study_id IR-Efimov-000002:
The tetramer set has additional subjects p1426 and p1507.
The tetramer set has additional sample/repertoires for subjects p1448 (2 files), p1484 (3 files), and p1495 (4 files).
New samples/repertoires for p1448: p1448_exp_YLQ_neg_alpha, p1448_exp_YLQ_neg_beta
New samples/repertoires for p1484: p1484_exp_RLQ_neg_alpha, p1484_exp_RLQ_neg_beta, p1484_exp_RLQ_pos_beta
New samples/repertoires for p1495: p1495_exp_RLQ_neg_alpha. p1495_exp_RLQ_neg_beta, p1495_exp_RLQ_pos_alpha, p1495_exp_RLQ_pos_beta
The IFN set has new subjects p1434, p1448, p1449, p1465, p1477, p1480, p1481, p1482, p1486, p1494, p1495, p1531, p1545, and p1551. Of these, subjects p1465, p1477, p1481, p1482, p1486, p1531, p1545, p1551 are new subjects relative to IR-Efimov-000001 (i.e. they were not previously included in the IFN or tetramer sets).
2021-01-26
All studies: Study title (study_title) was updated for all studies to include the last name of the first author to facilitate searching. Field Study Contact was added (emerging AIRR field) that lists a contact person for each study was added. Lab name and Lab address were updated for consistency. Publication links were updated in some cases to reflect preprint to publication status.
For project PRJNA648677 (Kim et al.) the sex of the subject was loaded as Male/Female whereas the correct controlled vocabulary should be male/female (all lowercase). This has been fixed.